
Research
We study how development shapes genetic inheritance, using plant systems to address questions with broad implications for biology, evolution, and crop improvement. Current projects include:
Gene regulation and selection in the haploid phase
Flowering plants alternate between multicellular haploid and diploid generations. Because pollen actively transcribes its haploid genome, recessive alleles that are masked in diploid tissue are directly exposed to selection, making the haploid phase an effective filter on genetic variation. We use single-cell genomics and functional approaches to follow haploid gene expression from meiosis through pollen maturity. A central question is: how much of pollen development is under haploid genetic control, and how much relies on transcripts inherited from the diploid parent?
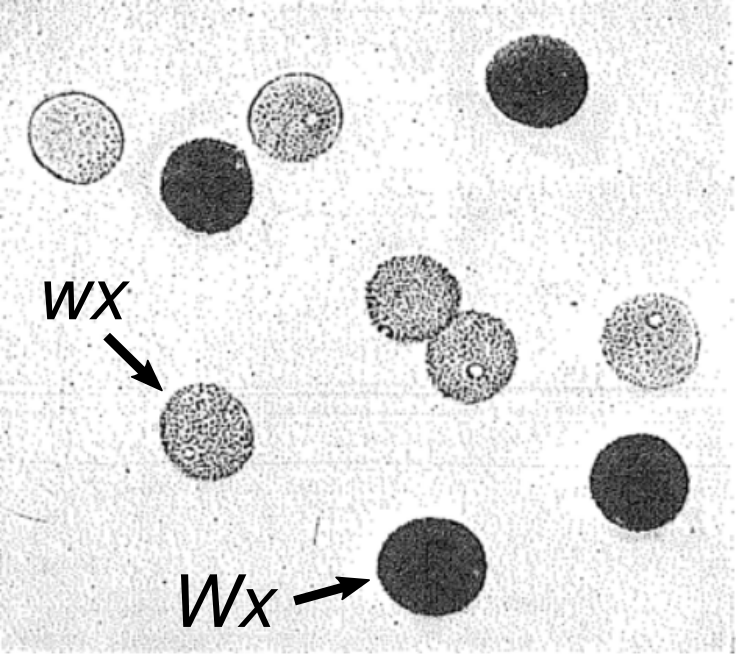
Fig. 1. Haploid genotype connects to haploid phenotype in pollen. Pollen grains from a single rice plant heterozygous for the waxy gene (Wx/wx), stained with iodine. Dark grains carry Wx; light grains carry wx. These same alleles distinguish non-sticky rice from sticky rice. Adapted from Parnell (1921).
The sporophyte-to-gametophyte transition. Using allele-specific single-cell RNA sequencing in maize, we found that diploid-derived transcripts persist for ~11 days after meiosis, followed by a rapid, genome-wide shift to haploid gene expression at pollen mitosis I — a sharp transition we named the sporophyte-to-gametophyte transition (SGT) (Nelms and Walbot, 2022). Genes activated after the SGT experience stronger purifying selection than those expressed only before it. We are now asking if the SGT is conserved across plant species, and whether variation in its timing affects the scope of haploid selection.
Mechanisms of haploid genome activation. What triggers the shift from diploid-derived to haploid gene expression? In collaboration with the Gent lab at UGA, we showed that DNA demethylation by two DNA glycosylases is required for expression of ~60 pollen genes that together account for ~11% of the poly(A) transcript pool in mature pollen (Zeng*, Somers* et al., 2024), identifying targeted demethylation as a key regulator of haploid gene expression. We are now investigating the broader chromatin-level changes that precede and accompany the SGT.
Haploid genetics at scale. Pollen offers a unique opportunity for large-scale genetics: each maize plant produces millions of pollen grains whose genotype is directly linked to phenotype. We are developing pooled loss-of-function screens using transposon-tagged populations and flow cytometry, creating a platform for genetic discovery at a scale that is difficult to achieve in the diploid plant.
Multicellular development and the spread of new mutations
Mutation is an inevitable fact of life. With deep sequencing, it is now clear that multicellular organisms accumulate tens to hundreds of mutations per cell over a lifetime. What determines whether a mutation spreads through the body or remains confined? We are leveraging unique features of the plant life cycle to build a quantitative, population-genetic understanding of how development shapes mutation fate.

Fig. 2. Mutations during development spread clonally, their reach governed by reproducible rules of growth and cell division. Left: a color mutation in a single cell gave rise to a stripe of differently pigmented tissue in this flower. Right: patches of white tissue mark cell lineages that lost chlorophyll production. Plants are particularly powerful for studying mutation spread — mutations from active transposons and other unstable elements can be readily followed visually and by sequencing.
Population genetics of a multicellular organism. New mutations are challenging to identify because they can be rare and difficult to distinguish from sequencing errors. We have established multiple approaches to track mutations within plants and across generations: using transposons as a targeted counting system (Scherer et al., 2025), single-molecule sequencing (Meyer et al., 2025), and unstable minichromosomes that can be followed in space and time via microscopy (in progress). We found that although individual mutations arise stochastically, their aggregate frequency distributions are highly reproducible between plants — indicating that development shapes not just mutation rate per cell but how far mutations expand clonally. We are now combining population genetic modeling with developmental mutants to understand what features of multicellular organization allow mutations to spread — or prevent them from doing so.
The distributive germline. A genetic bottleneck amplifies whatever variants its founders carry, and the germline is no exception. When germ cells share a recent common ancestor, mutations in that lineage are transmitted to offspring at elevated frequencies regardless of their selective value. We have found that many organisms limit germline bottlenecks by maintaining multiple deeply diverged progenitor lineages — a strategy we term a distributive germline — so that no single mutation can dominate the next generation. Independent lineage-diversifying mechanisms appear across the tree of life: growth geometry in plants, asymmetric germ granule segregation in zebrafish, and early cell mixing in mammals. We are testing the model’s predictions using densely sampled tissue collections, microscopy-based lineage tracing, and mutants with altered meristem organization (Scherer et al., 2026).
Reprogramming the plant cell
Richard Feynman once said: “What I cannot create, I do not understand.” For cell fate, the test is whether we can induce a desired transformation ectopically. Ectopic expression of transcription factors (TFs) has already yielded powerful tools in plant biotechnology. We aim to extend this paradigm to new cell fate transformations, with a focus on inducing meiosis and pollen formation from somatic cells in culture. This would enable new genetic manipulations and dramatically shorten plant breeding cycles.
A systematic survey of TF function in maize. The maize genome encodes ~3,000 TFs, and those capable of inducing any given cell fate transformation are almost entirely unknown. We have developed a robotics-assisted platform in which individual TFs are expressed transiently in maize leaf protoplasts in 384-well plates, with the full transcriptome-wide response measured by RNA-sequencing — converting a combinatorial search problem into a systematic survey of TF function. One third of the planned survey (656 of 2,112 TFs) has been completed, with the large majority of TFs inducing reproducible and distinct transcriptome responses. Candidates for regeneration and pollen development have been identified and are moving into secondary functional assays. The complete dataset will also serve as a resource for gene regulatory network inference across maize. For more information, see the TF Atlas project page.
Functional screening to functional validation. TFs that induce gene expression programs in protoplasts move into lower-throughput secondary assays testing for physiological responses — for example, whether candidate regeneration TFs can induce ectopic cell divisions in immature embryos or seedling leaves. Promising candidates are then pursued in full transformation and regeneration protocols, with optimization of TF combinations, promoters, and source tissues. This staged pipeline makes the discovery process tractable even for ambitious targets like meiosis induction, where no prior candidates exist.